The Regulation and Governance of Medical Devices in Scotland
The regulation of medical devices in Scotland is complex and often poorly understood. In recent years, there has been considerable public concern over the safety of some medical devices and an increase in the number of medical devices subject to recall. This briefing looks at the regulation and governance of medical devices in Scotland. It also looks towards the future of medical device regulation in light of the UK’s anticipated departure from the EU regulatory framework following Brexit.
Executive Summary
Medical devices are used in the diagnosis, treatment and management of a wide range of diseases and conditions. In the UK, one in twenty-five people has an implanted medical device1.
Medical devices can be divided into two broad categories, medical devices and in vitro diagnostic medical devices, and classified by risk.
The regulation of medical devices is reserved to the UK Parliament, which implements EU directives on medical devices. In 2017, the EU brought in new regulations on medical devices.
The UK regulator, the Medicines and Healthcare Products Regulatory Agency (MHRA), covers all medicines and medical devices used in the diagnosis and treatment of illnesses. Under the current UK Regulations, manufacturers must report adverse events involving medical devices to the MHRA. In the Scottish context, Health Facilities Scotland (HFS) assists the MHRA in providing technical and operational support to the Scottish Government Health and Social Care Directorate and NHSScotland.
Scottish companies involved in research and development of medical devices aim to develop devices from concept to being ready for the market2. Some commentators2 report that the high pace of development and low adoption rates within NHSScotland, and Scotland more generally, creates a challenging environment for medical device companies.
The Health Innovation Assessment Portal (HIAP) is managed by NHS National Procurement. The HIAP is the first step in a national process that is being developed to provide health innovators with feedback from NHSScotland. The portal can canvass and receive feedback on submissions from across a broad cross-section of NHSScotland.
Industry experts42 believe a more transparent procurement strategy is needed and that the centralisation of procurement and monitoring of medical devices would lead to greater control. In 2018, the NHS National Services Scotland Procurement Strategy was published. The strategy encouraged a closer working relationship between health boards and National Procument NHSScotland, but did not suggest a centralised approach.
Under the new European regulations, any medical intervention that uses digital automation to generate information about patients is now considered to be a medical device and must be CE marked. The procurement strategy has resulted in some technologies being removed from the NHS. However, recent Scottish Government strategies reflect the move to more collaborative and increasingly digital models of healthcare. As such, governance is required to help include digital technologies in the healthcare sector to comply with these regulatory changes.
The UK is due to leave the EU in March 2019, and this may leave the UK and EU open to a regulatory divergence6.
What are Medical Devices?
Medical devices are used in the diagnosis, treatment and management of a wide range of diseases and conditions. They vary significantly in complexity and application and include many things from bandages to magnetic resonance imaging (MRI) scanners. In the UK, one in twenty-five people have an implanted medical device1 and over 500,000 different types of medical devices are available worldwide 2.
Definitions of medical devices are typically wide-ranging and often based on what they are not, rather than what they are i.e. products used for healthcare purposes that are not medicinal in nature. Such definitions make it difficult to be certain of where medical devices end and other technologies, such as medicines, begin.
The European Union (EU) defines a medical device as:
Any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
• Diagnosis, prevention, monitoring, treatment or alleviation of disease
• Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap
• Investigation, replacement or modification of the anatomy or of a physiological process
• Control of conception
Which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means.
From Article 1 of EU Directive 93/42/EEC.
What Types of Medical Devices are there?
Medical devices can be divided into two broad categories.
1. Those which can be used to diagnose, treat or monitor patients, including apps and software (see table 1 for examples), and
2. In vitro diagnostic medical devices (IVDMDs) which are used to examine specimens outside the body. For example, pregnancy tests and blood glucose monitors.
| Function | Examples |
|---|---|
| Diagnosis or treatment of disease | Diagnostic laboratory devices, x-ray machines, MRI scanners, vascular catheters, dressings, surgical instruments, syringes, hip replacement implants, stand alone software for diagnosis. |
| Monitoring of patients | Electrocardiogram (ECG), pulse oximeters. |
| Critical care | Infant incubators, blood gas analysers, ventilators, vascular stents. |
| Improving the function and independence of people with physical impairments | Hoists, orthotic and prosthetic appliances, pressure care devices, walking aids, wheelchairs. |
| Community-based healthcare | Dressings, domiciliary oxygen therapy systems, catheters. |
| Emergency services (ambulances) | Stretchers, trolleys, defibrillators. |
Source: (MHRA, 2015)
How are Medical Devices Classified?
Medical devices are classified by risk. Classes of medical devices are based on contact time, invasiveness and whether they are active or inert if placed in the body1. A set of criteria are used to determine a device's classification and this influences the level of regulatory control. Table 2 shows the different classes of medical devices, the corresponding risk levels and examples of devices in each class.
| Class | Risk | Examples |
|---|---|---|
| I (basic) | Low | Reusable surgical instrument, non-sterile gloves. |
| I (sterile) | Low | Sterile dressings (non-medicated), sterile gloves. |
| I (measuring) | Low | Volumetric urine bag. |
| IIa | Medium | Surgical blades, hypodermic needle, suction equipment. |
| IIb | Higher | Ventilators, orthopaedic implants, radiotherapy equipment. |
| III | Highest | Prosthetic joints, coronary stent. |
Source: (Matherson et al., 2013)
How are Medical Devices Regulated?
The regulation of medical devices is reserved to the UK Parliament. Medical devices are currently regulated under the:
These seek to ensure that:
Medical devices are designed and manufactured so that they will not compromise the safety or the clinical outcomes of patients.
Medical device manufacturers must display a Conformité Europeénne (CE) marking to show that they are safe and fit for their intended purpose.
CE marking must be awarded by an EU accredited private organisation, known as Notified Bodies. Notified Bodies are responsible for the evaluation of the submitted clinical data by the manufacturers.
Once a CE mark has been awarded in one country it enables access to the entire EU market.
In 2017, the EU brought in new regulations on medical devices. These differentiate between medical devices and vitro diagnostic medical devices (IVDMDs)1. The new regulations are:
Changes to EU regulations
The new Regulations aim to make a number of improvements to modernise the current system including:
Stricter control for high-risk medical devices.
Stricter control over how Notified Bodies work.
Improved transparency and traceability of medical devices through the establishment of an EU database.
The introduction of an 'implant card' containing information about implanted medical devices for a patient.
Stricter rules on what constitutes clinical evidence in the pre-market stage.
Stronger post-market surveillance for manufacturers.
Improved coordination mechanisms between EU countries in relation to vigilance and market surveillance2.
The new regulations on medical devices have a three year transition period, whereas the IVDMDs regulations have a five year transition period. During the transition period, devices can be placed on the market under the current EU Directives, or the new Regulations (if they fully comply with the new Regulations)3.
The Medicines and Healthcare products Regulatory Agency (MHRA)
The Medicines and Healthcare products Regulatory Agency (MHRA) covers all medicines and medical devices used in the diagnosis and treatment of illnesses. It is responsible for monitoring the efficiency of products and responding quickly when safety concerns are raised. It aims to achieve compliance through the provision of advice and guidance and to maintain standards of quality and safety through a risk-based inspection programme. The MHRA also has the power to withdraw a product from the market and prosecute the manufacturer or distributor. 1.
MHRA Safety and Quality Standards Monitoring Processes
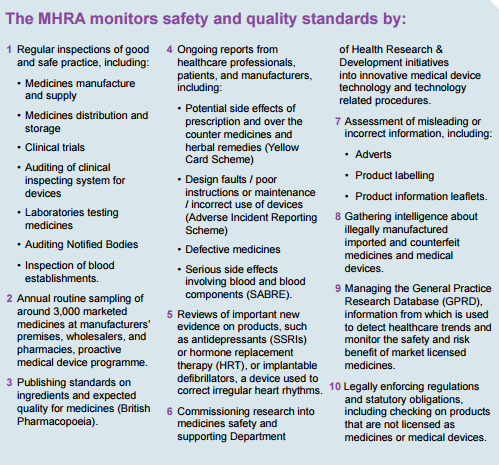
(MHRA, 2012)
When a product is suspected or known to be faulty, the MHRA works with the manufacturer and wholesaler to agree the most appropriate action to take. In serious circumstance, the product has to be recalled and taken out of the supply chain. The MHRA oversees:
Field Safety Notices (FSNs) - sent out by medical device manufacturers or their representatives outlining actions they are taking in relation to a product.
Medical Device Alerts (MDAs) - issued by the MHRA to communicate safety information to device users in health and social care.
The MHRA also operates the Yellow Card Scheme which monitors the safety of medicines and devices in the UK. Reports can be made by healthcare professionals and patients about safety concerns on products via the Yellow Card Scheme.
Reporting of adverse incidents
Under the current UK Regulations, manufacturers must report adverse events to the MHRA through the Manufacturers’ Online Reporting Environment (MORE) which allows for vigilance reports, and responses, to MHRA incident investigations to be submitted.
Reporting and monitoring arrangements for professionals working in the Scottish health and social care system are set out in CEL 43 (2009). In the Scottish context Health Facilities Scotland (HFS) provides technical and operational support to the Scottish Government Health and Social Care Directorate and NHSScotland bodies in relation to aspects of healthcare facilities.
HFS operates an incident reporting system as part of NHSScotland. This reporting scheme is operated by the Incident Reporting and Investigation Centre (IRIC). IRIC offers a comprehensive approach for reporting incidents involving medical devices, social care equipment, laboratory equipment and estates and facilities equipment.
HFS works closely with MHRA, and will notify it of each adverse incident reported in Scotland and the results of any investigation, and vice versa 1. The outcome of an investigation may also lead to the dissemination of safety alerts and notices which are shared with stakeholders in Scotland and the rest of the UK. Safety alerts help NHS boards and local authorities to put controls in place locally to minimise the chance of recurrence.
Whilst manufacturers have to report adverse incidents to the MHRA they do not have to report them to IRIC. This has led to underreporting. At the 2018 annual HFS-IRIC conference IRIC Manager, Innes Connor, stated that the data received by IRIC did not give a full overview of the real numbers of adverse incidents that had occurred.
To ensure that safety alerts are acted upon appropriately by health boards and local authorities, a pilot scheme for monitoring of safety alerts is being carried out by HFS. A recent announcement by the Scottish Government indicates that funding is available for IRIC to develop guidance for the management of medical devices and equipment. The guidance is due to be published in 20202.
High Profile Medical Device Incidents
In recent years, there has been a number of high profile cases relating to problems with medical devices, including poly implant prothèse (PIP) breast implants, metal-on-metal hip implants and urogynaecological mesh implants.
PIP (Poly Implant Prothèse’s) Silicone Breast Implant
PIP silicone breast implants were withdrawn from the UK in 2010 following evidence that the company had deliberately concealed the use of non-approved industrial-grade silicone.
National Cancer Institute. (2008). Hand holding a breast implant which is a flexible sac filled with silicone gel [Accessed 23rd May 2018. Retrieved from Wikimedia Commons [accessed 23 May 2018]
A 2010 SPICe Briefing for the Public Petitions Committee stated that, as of 2010, 10,000 people in the UK received breast implants in the UK each year2. The MHRA first received reports of potential problems with PIP implants in 20023. Information on adverse events was given to the manufacturers from 2003. Following an inspection of the French-based company by national health authorities (Agence française de sécurité sanitaire des produits de santé), a ban on PIP implants in the UK was announced in March 2010.
The ban resulted from concerns about the use of an unapproved filler. Following testing it was concluded that the company had been using non-medical grade silicone, similar to that which is used in mattresses, to fill their implants4. PIP implants had double the rupture rate of other implants but were not found to be toxic or carcinogenic. The MHRA received 269 adverse incident reports relating to PIP silicone implants between 2001 and 2009, including a case of premature rupture of both implants in the same patient3.
Timeline of events
Timeline of events in the PIP Case

The Telegraph. (2012). Breast implant scandal: timeline of how events unfolded. Retrieved from https://www.telegraph.co.uk/news/health/news/9146338/Breast-implant-scandal-timeline-of-how-events-unfolded.html [accessed 28 June 2018]
Stakeholder action
In July 2010, the MHRA commissioned analysis to provide a preliminary evaluation of the filler material used in the implants1. The evaluation revealed the presence of silicone compounds in addition to traces of organic and inorganic impurities. Based on subsequent laboratory analysis the MHRA concluded, in June 2012, that this ‘did not raise any concerns regarding risks to human health’ 2.
The Lancet. (2009). Hand holding PIP breast implant. Retrieved from Doi: https://doi.org/10.1016/S0140-6736 [accessed 29 May 2018]
The Expert Group on PIP, chaired by Sir Bruce Keogh, was appointed by the UK Department of Health (DOH) in 2012. The DOH review of PIP aimed to determine whether the actions of the MHRA and the UK Government could have reasonably prevented or uncovered the problem earlier. It reported that the primary concern was, “The fact that PIP deliberately concealed their use of a non-approved filler material has rightly triggered questions about how this can have happened, and how it remained undetected for such a long period”2.
The report concluded that MHRA had fulfilled its obligations reviewing and responding to the incidents reported, but that investigations were hampered by a lack of reliable and comparative data on similar products. It noted conclusions had been drawn by the MHRA from incomplete data that had been filtered through a fraudulent manufacturer. The report praised the actions of the MHRA, such as ordering in house testing of PIP when French toxicology reports were taking too long. The report suggested that more accurate data on adverse incidents was needed, that it should be provided promptly and that information sharing across international boundaries should be comprehensive, timely and accurate.
DePuy’s Metal on Metal (MoM) Hip Prostheses
National Institutes of Health. (2006). An anteroposterior (A-P) X-ray of a pelvis showing a total hip joint replacement. Retrieved from Wikimedia Commons [accessed 23 May 2018]
The ASR™ MoM hip prostheses first became first available in Europe under the clause of ‘substantial equivalence’. The clause allowed fast-track market accreditation of the device on the basis that the implant was similar to one already on the market and that, therefore, it was not required to have its own safety and efficiency clinical data.
Problems with the device were first raised in the UK in 2008, at orthopaedic conferences and in journal publications. The device was reported to cause muscle damage, bone damage and metal toxicity. In 2009, the annual report of the National Joint Registry (NJR) recorded comparatively higher rates at which the hip replacement had to be replaced for the ASR™ hip.
Despite these findings, official notices about problems with the DePuy ASR™ were “[…] limited to guidance on positioning during surgery”2. In August 2010, DePuy issued a global recall of all ASR™ hip systems3. Following this recall, in 2013 the MHRA issued a Medical Device Alert (MDA), advising that patients with MoM hips were to have regular medical interventions to monitor the build-up of ions in the blood4. In June 2017 this MDA was updated to indicate that ultrasound and MRI scans should also be used in decision-making 5.
Timeline of Events
Timeline of events in the MoM hip case

The Telegraph. (2017). Hip Implant Maker Alerted Safety Fears Surgeon Told Borders. Retrieved from https://www.telegraph.co.uk/news/2017/04/11/hip-implant-maker-alerted-safety-fears-surgeon-told-borders/ [accessed 23 May 2018]
Stakeholder Action
A number of organisations played a significant role in the post-market medical implant surveillance and regulation of the ASR™ hip, including: The manufacturer (ASR), the MHRA, (which appointed a Metal-on-Metal Expert Advisory Group), the National Joint Registry (NJR), the British Orthopaedic Association and the British Hip Society. It is unclear whether De Puy knew about issues with the device before removing it from the market in 20111. Prior to ASR™ withdrawal in 2010, neither the BOA nor the BHS published formal guidance or recommendations on the accumulating failure data for the ASR™. The BOA did engage with MHRA to raise the regulator's awareness of the problems with the ASR™ hip. BOA also funded to investigate the poor clinical performance of the ASR™1. However, they noted that more could have been done to raise awareness and that professional bodies should ‘be proactive if similar problems arise again’3. The primary driver for uncovering the failures of the ASR™ hip was surgeons publishing their data in journals and/or conferences.
There has been no formal DOH review of the ASR™ hip. However, a House of Commons Science and Technology Committee report, titled: Regulation of medical implants in the EU and UK, included sections on MoM Hips.
This case shows that medical devices can fail in ways that are very technical and that it is sometimes difficult to determine what aspect of a device causes failure. It also illustrates how replacing a harmful device is not always clinically justified, in cases where the risk of surgery outweighs the risks presented by the device.
Polypropylene Urogynecologic Surgical Mesh Implants
Mesh is a term used to describe a range of synthetic or biological implants that can be used to provide additional support when repairing weakened or damaged tissue1. Mesh implants can be used in a variety of surgical interventions across both genders, for example, for hernia repair. However, concerns have been raised over the use of mesh to treat pelvic organ prolapse (POP) and stress-urinary-incontinence (SUI).
Concerns about mesh were first raised in the United States in 19992. A SPICe Briefing for the Public Petitions Committee reported that,"[...] in terms of surgery for POP there is a 20% to 30% failure rate from primary prolapse surgery and women may need second and subsequent procedures to address prolapse recurrence."3
Polypropylene Mesh Adjust Single - Incision Mid Urethral Sling
Polypropylene Mesh Adjust Single - Incision Mid Urethal Sling. (n.d.) Retrieved from Creative Commons [accessed 23 May 2018]
It is clear that a number of women have suffered serious, life changing complications following TVM surgery. It is also evident that many women have benefited from these procedures. However, due to the way these procedures are coded, it is not possible to provide accurate data on the number of mesh procedures where complications have occurred. This lack of information, allied with the fact that adverse events have been under-reported, has led to opinion being divided on the safety of transvaginal mesh procedures. […] No procedure is without risk and therefore many people, including the broad clinical community, consider that polypropylene mesh should continue to be used in some circumstances as it presents an acceptable level of risk […]1.
Timeline of Events in the Polypropylene Urogynecologic Surgical Mesh Case
A Timeline of Events in the Polypropylene Urogynecologic Surgical Mesh Case has been curated by the Centre for Evidence-Based Medicine at the University of Oxford.
Stakeholder action
The former Cabinet Secretary for Health and Wellbeing, Alex Neil MSP, met with a group of women adversely affected by the use of mesh in May 2013. Following this, a Scottish Government working group was established to address the issues affecting women who have undergone TVM surgery. The working group recommended developing care pathways for women experiencing complications and to improve the consent processes surrounding TVM surgery.
In 2014, the Scottish Government requested the suspension of the use of mesh1 and the Chartered Society of Physiotherapy called for greater access to physiotherapists at this time2. In 2015, the Scottish Government published its interim report, ahead of the final report of the Scottish Government inquiry on mesh. Two campaigners, Olive McIlroy and Elaine Holmes, resigned from the group in March 2017, along with urogynaecologist Wael Agur, stating: “The report did not adequately warn surgeons and patients against the serious risks with the transobturator mesh tape”3.
In March 2017, the final report from the Scottish Government was published. This report recommended that mesh should not be used over native tissue repair4. It advised that mesh should be used as a second line therapy, but not in the first instance. It also recommended that:
Mesh must not be offered routinely to women with pelvic organ prolapse.
Reporting of all procedures and adverse events should be mandatory, in line with the guidance from the General Medical Council.
Extra steps should be taken to ensure that patients have access to clear, understandable advice to help them make informed choices.
In the case of surgical treatment for stress-urinary-incontinence, all appropriate treatments should be available.
Training should be improved for clinical teams involved in transvaginal mesh.
More research was needed into the safety and effectiveness of the products.
A new oversight group should be established.
In May 2017, Health Improvement Scotland (HIS) established a Mesh Implants Oversight Group and commissioned an independent evaluation of the initial inquiry into mesh.
Throughout 2016 and 2017, the National Institute for Health and Care Excellence (NICE) updated its advice on mesh. The NICE report of 2017 states mesh for vaginal wall prolapse should only be used in the context of research. This updated advice says that current evidence on the safety of the procedure shows there are serious, but well recognised safety concerns. The evidence for long term efficacy is inadequate in quality and quantity. Therefore, the procedure should only be used in the context of research5. The Royal College of Surgeons and Gynaecologists published new decision making materials regarding mesh in 2017.
Future investigations into mesh include a DOH review which was launched in February 2018. This review will focus on three NHS treatments:
TVM
Primodos (a home pregnancy test kit)
Sodium Valproate (anti-epileptic drug)
In 2018, UK Government Health and Social Care Secretary, Jeremy Hunt MP, has announced a review into how the health system responds to reports from patients about side effects from treatments6.
Innovation and Governance in the Medical Devices Sector
Scotland is home to a large and vibrant medical devices industry encompassing more than 250 companies, with over 9,000 people employed in the sector1. Scottish companies involved in research and innovation in medical devices aim to develop devices from original concept (technology readiness level 1) to be ready for market (technology readiness level 9)2. Some commentators2 report that a high pace of innovation and change, but low adoption rates within NHSScotland creates a challenging environment for medical device companies.
The table below outlines the technology readiness levels as well as the funding streams that support manufacturers in these various stages.
Medical Device Innovation Pathway
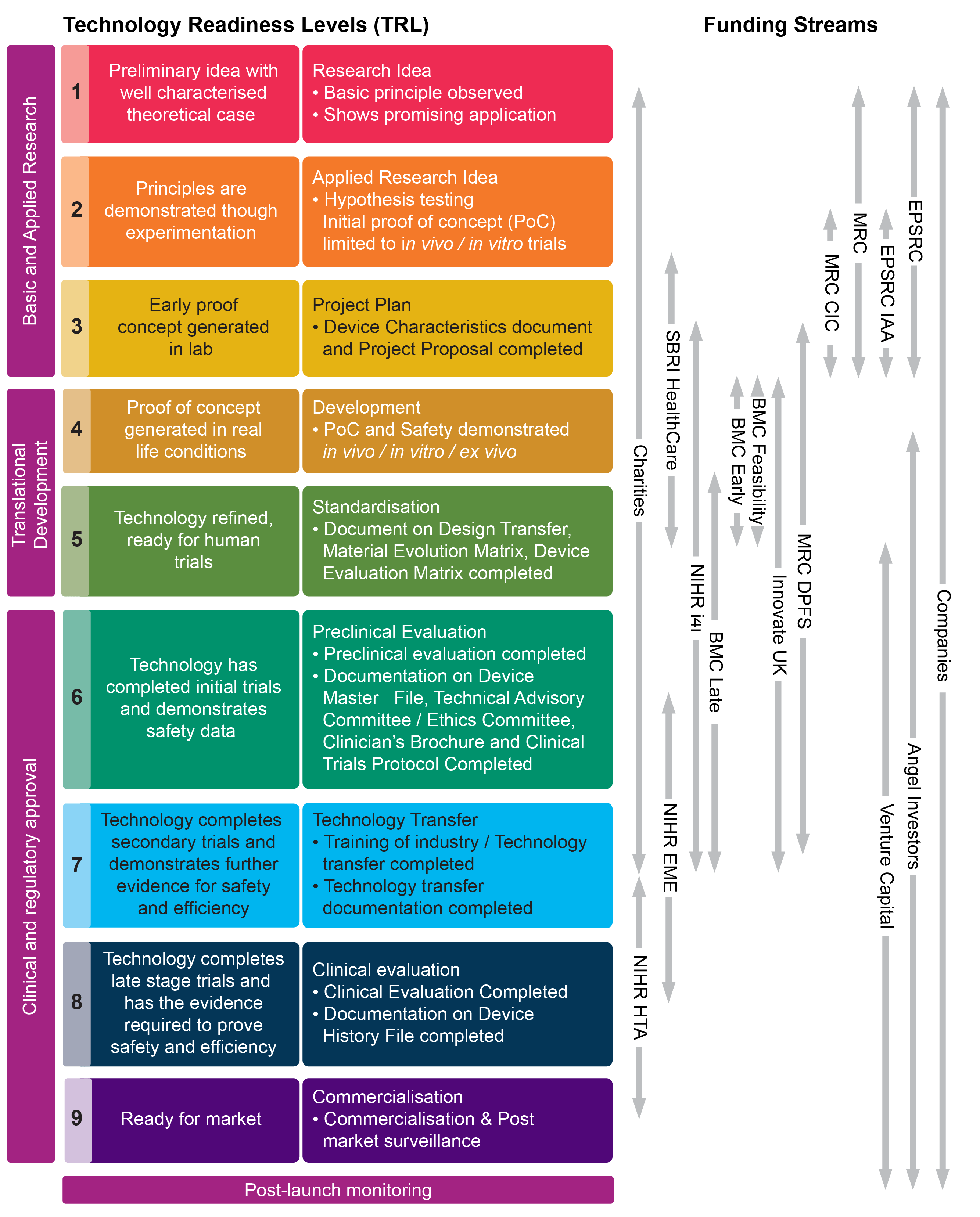
Nuclear Decommissioning Authority. (2014). Guidance on Technology Readiness Levels. Retrieved from https://www.gov.uk/government/news/guidance-on-technology-readiness-levels [accessed 13 June 2018]
The Health Innovation Assessment Portal
The Health Innovation Assessment Portal (HIAP) is an online platform designed as a single point resource to develop stronger relationships between the NHS and industry. HIAP is managed by NHS National Procurement. The HIAP is the first step in a national process that is being developed to provide health innovators with feedback from NHSScotland. The portal has the ability to canvass and receive feedback on submissions across a wide cross-section of NHSScotland.1
Companies can use the portal to get help to develop ideas, products and technologies that may be of potential use to NHSScotland. NHSScotland can use the portal to assess how an innovative medical device might support NHSScotland's strategic aims, what the associated costs and benefits would be. Ideas can be assessed by experienced and qualified healthcare professionals who have the opportunity to provide constructive criticism and feedback2.
Representation of a Medical Device Innovation Cycle
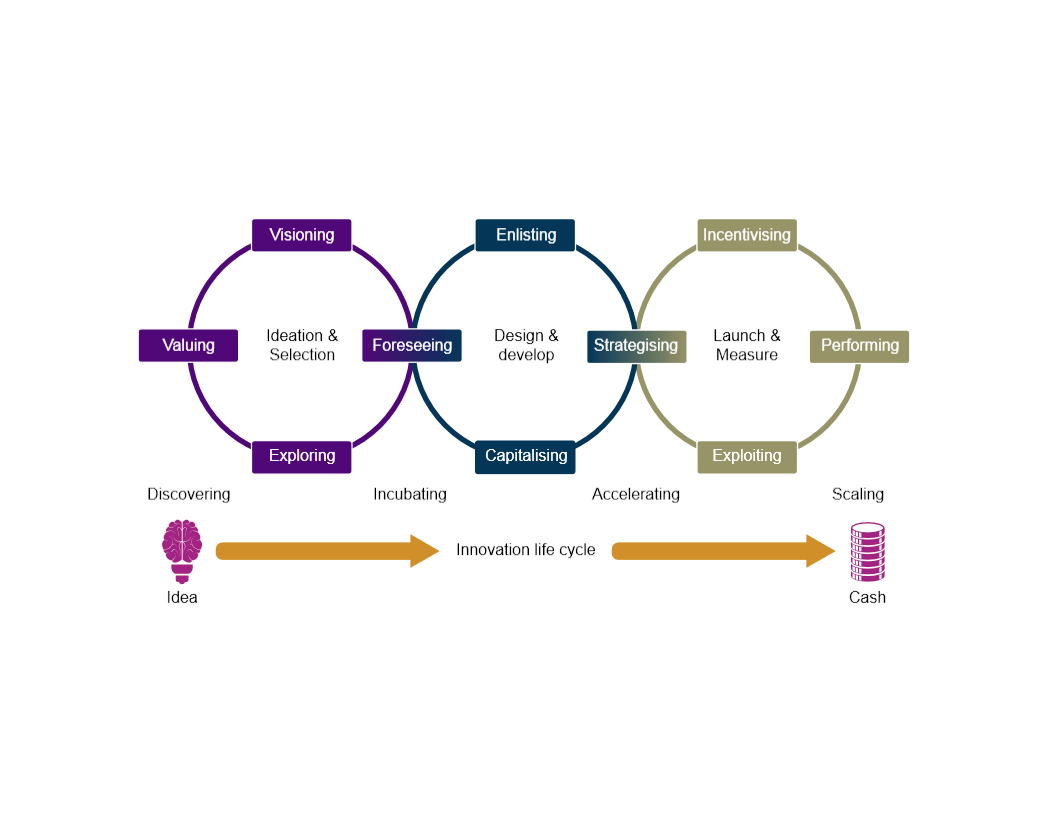
PwC. (2011). Representation of a Medical Device Innovation. Retrieved from http://usblogs.pwc.com/emerging-technology/powering-the-innovation-life-cycle/ [accessed 23 June 2018]
The HIAP, enables innovators to upload their ideas and solutions under six headings which enable innovators to establish whether an innovative idea is a good fit for investment. These are:
General information
Benefits
Market readiness and evidence
Commercial information
Strategic fit
Other information
HIAP response types
Further information sought
Assessors need more detail on particular aspects of the submission.
Incremental or “Me Too”
From the evidence provided there is significant overlap with existing applications or technologies (whether or not currently used by NHS).
No likely fit
While the solution may (or may not) have interest it is not clear how the adoption would support the strategic aims of NHS Scotland.
Signposting
To other organisations that can provide advice and support.
Samples requested
If the submission is sufficiently developed samples may be requested for further assessment.
Progression to formal review assessment
The submission has clear potential but requires further consideration4.
Following this process manufacturer must take the device to prepare for the market. This is known as the premarket approval process (discussed below).
Pre-market approval
Before being marketed, a device must receive a CE mark which indicates that it complies with the relevant European directives. Compliance is reviewed in accordance with clinical data that is provided by the manufacturer. Clinical data can consist of the evaluation of the relevant scientific literature relating to the safety, performance, design characteristics and intended purpose of the device and/or the evaluation of the results of all the clinical investigation1.
The box below describes the process for randomised control trials and the challenges medical devices pose to this method of evaluation.
Randomised control trials (RCTs)
Medicines are typically evaluated using randomised control trials (RCTs). Medical devices tend to be evaluated using statistical methods for the analysis of clinical and economic data. This is, in part, due to lower evidence hurdles for medical devices than medicines.
There are a number of challenges in designing RCTs for medical devices.
With high-risk surgical innovations, such as implanted cardiac devices (ICDs), the use of RCTs for the assessment of safety and efficacy is often considered unethical 2.
The safety and effectiveness of medical devices are, in part, determined by the clinician's skill and patient selection. Training in the use of a medical device has been found to affect outcomes.
It may be impractical to repeat clinical trials for every design modification of a device.
A number of alternatives to RCTs exist for medical devices. For example, parallel group non-randomized studies or controlled interrupted-time series studies which examine specifically selected patients over a period of time. However, some academics have indicated that the use of randomised, double-blinded, sham-controlled medical device trials might be a better alternative.
The procurement and adoption of medical devices
Scottish public sector procurement operates under the following legislative framework:
NHSScotland's national procurement strategy is set out in the National Procurement Interim Strategy (2016-2018). This aims to promote the "four procurement pillars":
To provide continuity of supply
To provide value to the bottom line
To maintain effective governance
To encourage and stimulate economic development1
The NHS National Procurement works under the Scottish Model of Procurement. This aims to engage with service users and suppliers to facilitate a more comprehensive approach to national health procurement in Scotland.
The procurement of medical devices in NHSScotland is currently conducted at national, regional and local levels. Commentators have identified two main concerns with this approach:
The process is piecemeal.
There is no robust way of keeping track of devices, maintenance and calibration at a national level.2
Medical devices are more difficult to manage than medicines in terms of procurement as there are many more devices on the market that medicines. Medical devices are also constantly subject to innovation and change.
Market assessment is conducted by National Procurement NHSScotland in conjunction with an advisory panel made up of health board representatives. Some people believe that NHSScotland's procurement strategy lacks transparency3 which impacts on adoption3.
Some people believe a clearer procurement strategy is needed and that the centralisation of procurement and monitoring of medical devices would lead to greater control. In 2018, the NHS National Services Scotland Procurement Strategy was published which encouraged a closer working relationship between health boards and National Procument NHSScotland but did not suggest a centralised approach. However, the NHSScotland Procurement Transformation Programme5 aims to have the "Seamless eProcurement Technology Infrastructure in Place" by 2018.
Post-market surveillance
Post-market surveillance allows safety concerns about medical devices to be raised by manufacturers, patients or clinicians. Post-market testing is important due to the often long life cycles of medical devices1.
Medical devices have a cyclical model of innovation and, often, manufacturers continue to test medical devices after they have reached the market. Under the new EU regulations a post marketing surveillance (PMS) plan is required as part of the submission of evidence for market surveillancei. This will be carried out by the manufacturer. A PMS plan must include:
information on serious incidents, including information from Periodic Safety Update Reports (PSUR) , and Field Safety Corrective Actions (FSCAs)
records on non-serious incidents and data on any undesirable side-effects
information from trend reporting
relevant specialist or technical literature, databases and registers
information provided by users, distributors and importers including feedbacks and complaints
publicly available information about similar medical devices2
PMS is a reactive process and, as such, it cannot on its own prevent the risk of harm caused by medical devices, although, it can help in the earlier identification of problems.
Future challenges for the regulation and governance of medical devices
Mobile Health, software and applications as medical devices
Under the new European regulations, any medical intervention that uses automation to generate information about patients is considered a medical device and must be CE marked. This has resulted in some technologies being removed from the NHS. For example, a spreadsheet with mathematical functions that was used to calculate patient hydration is no longer used because it does not have CE approval1.
The use of Mobile Health (MHealth) is increasing and 2 some NHS Trusts in England have even used guidance for the development of mobile health apps. MHealth can be used for a wide range of purposes, including for communication between people and health systems, health monitoring and access to information such as health records and decision support2.
Digital health and MHealth technologies further complicate the landscape of medical devices regulation because, although they are regulated in the same way as medical devices,the risks they pose to patients is very different to that of traditional medical devices.
Increasingly, MHealth is being combined with other innovations, for example, the internet of things (IOT)i, artificial intelligence or machine learning in order to better maximise the data collected from mobile health applications. In the future, these hybrid devices will play an important role in proving healthcare.
| Description | Examples |
|---|---|
| Mobile applications that are an extension of one or more medical device or display, store, analyse or transmit patient-specific medical device data. | Remote display of data from bedside monitors.Control of inflation/deflation of a blood pressure cuff.Control of the delivery of insulin by an insulin pump. |
| Mobile applications that transform the mobile electronic device into a medical device by using attachments, display screens, or sensors. | Attachment of a transducer to a mobile electronic device to function as a stethoscope.Attachment of a blood glucose strip reader to a mobile platform to function as a glucose meter.Attachment of electrocardiograph (ECG) electrodes to a mobile platform to measure, store, and display ECG signals. |
| Mobile applications that allow the user to input patient-specific information, output a patient-specific result, diagnosis, or treatment recommendation to be used in clinical practice or to assist in making clinical decisions | Mobile applications that provide a questionnaire for collecting patient-specific lab results and either: 1. compute the prognosis of a particular condition or disease; 2. perform calculations that result in an index or score; 3. calculate dosage for a specific medication or radiation treatment; or 4. provide recommendations that aid a clinician in making a diagnosis or selecting a specific treatment for a patient |
One of the main issues with the rising trend of virtual healthcare is that there has been a rising number of applications that produce lifestyle data. However, the data produced from many of these applications is considered not accurate enough for CE marking6.
Case study: Medical Device Software Based Clinical Decision Support for Diabetes in Scotland
The Scottish Government and NHSScotland have a national eHealth Strategy to promote quality of care, enabling shared decision-making with patients and integrating health and social care. One of the EHealth Good Practice Case Studies is based on Clinical Decision Support for Diabetes in Scotland. The Clinical Decision Support for Diabetes Case Study is a pilot run in Lothian and Tayside that aims to link My Diabetes My Way, Diasend and Scottish Care Information (SCI) Diabetes. My Diabetes My Way allows for secure access to patients' diabetes.
Diasend offers healthcare providers a cloud-based solution that stores all diabetes patient data centrally. The clinician treating the patient would receive analytics from Diasend and receive clinical decision support. The data from Diasend is automatically updated on SCI diabetes and My Diabetes My Way to help provide national epidemiological data for NHSScotland, and to provide a friendly user interface for diabetic patients to assist in patient-directed long-term condition management. Future aims of the project are to integrate new and emerging medical devices, like CGM or flash glucose monitoring to generate data and then to use regression-based analysis to generate predictive models, rather than rules-based models.
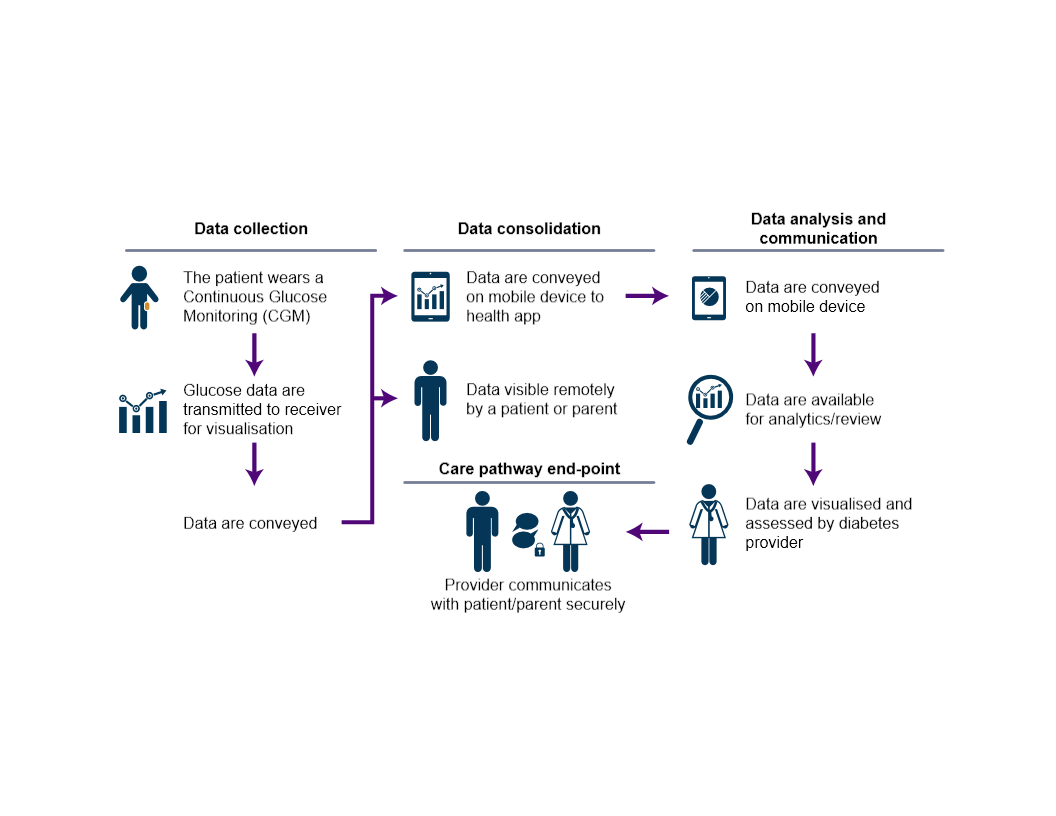
Scottish Government Strategies
Despite increased regulation surrounding digitally automated medical devices, the Scottish Government strategies reflect the move to more collaborative and increasingly digital models of healthcare. The Scottish Government's Digital Health and Social Care Strategy highlights the importance of the use of technology in healthcare.
Scotland's Digital Health and Social Care Strategy “Moving Forwards” We wish to empower citizens to better manage their health and well-being, support independent living and gain access to services through digital means. We know this is leading to a shift in the balance of care by using the tools and technologies that we are already increasingly using for all other aspects of our lives, and In order to achieve this at scale, we need to put in place the underpinning architectural and information governance building blocks for the effective flow of information across the whole care system that will enable the transformational ambitions of the Health and Social Care Delivery Plan, including public health and social care reform priorities.
The report by the Scottish Government's Expert Panel on Digital Health and Care in Scotland highlights the future potential of Mobile Health (MHealth) technologies. The report indicates an aim that every Scottish citizen will have a near real-time personalised view of their health information to which they can contribute. Ensuring that the technical infrastructures in health and care provide robust security protection for information is a real and recognised challenge as we move towards greater use of digital and outward facing systems, for example, with citizen portals and mobile apps. As such, national governance is required to help include digital health technologies in a way that complies with EU regulatory changes.
Brexit and the regulation of medical devices
As discussed earlier, new regulations on medical devices were adopted by the EU in 2017. These regulations have a transition period to 2020 for medical devices and 2022 for in vitro diagnostics medical devices.
However, the UK is currently due to leave the EU in March 2019 and this may leave the UK and EU open to a regulatory divergence1.
The Scottish Parliament's Health and Sport Committee undertook an inquiry into the issue and published its report in 201 and SPICe has published a briefing on the possible implications of Brexit on health and social care in Scotland.
Under the European Union (Withdrawal) Bill, it is proposed that the UK will continue to permit the sale of CE (European approved) items to avoid companies having to go through separate regulation processes.
Currently, once a medical device is approved it is available for sale anywhere in the EU. However, under EU regulations, that testing must take place within EU borders for a device to be supplied in the EU. For device manufacturers in the UK there is the possibility that devices will have to be double safety tested. A British Medical Association (BMA) Brexit Briefing on Medical Devices and Medical Regulation stated:
Adopting a divergent approach to licensing would lead to:
delayed access to new medicines and medical devices
In its inquiry, the Health and Sport Committee also heard about the potential impact of Brexit on the turnover of the UK's medical device industry, which is estimated to be worth £17 billion2.
In leaving the EU the UK will no longer have the same influence in shaping legislation, policy and regulatory procedures. However, there is scope for the UK to create its own system of certification 3.
Sources
Med Tech Europe. (2016). The European Medical Technology Industry – in figures. Retrieved from <a href="http://www.medtecheurope.org/sites/default/files/resource_items/files/MedTech_FactsFigures2016_20160105.pdf" target="_blank">http://www.medtecheurope.org/sites/default/files/resource_items/files/MedTech_FactsFigures2016_20160105.pdf</a> [accessed 20 June 2018]
In discussion with a representative from Strathclyde Institute of Medical Devices. (2018).
In discussion with a representative from Medical Devices Unit (NHS Greater Glasgow and Clyde). (2018, undefined).
Scottish Parliament Information Centre. (2018). Impact of leaving the European Union on health and social care in Scotland. Retrieved from <a href="http://www.scottish.parliament.uk/parliamentarybusiness/CurrentCommittees/107148.aspx" target="_blank">http://www.scottish.parliament.uk/parliamentarybusiness/CurrentCommittees/107148.aspx</a> [accessed 21 June 2018]
Chapman, A.M., Tayor, C.A., & Girling, A.J. (2104). Are the UK systems of innovation and evaluation of medical devices compatible? The role of NICE’s medical technologies evaluation programme (MTEP). [No description].
Masterson, F., & Cormican, K. (2013). Overview of the regulation of medical devices and drugs in the European Union and the United States. Therapeutic Innovation & Regulatory Scien, 47(6), 715-722. doi: https://doi.org/10.1177/2168479013500969
Medicines and Healthcare products Regulatory Agency. (2018). Medical devices: EU regulations for MDR and IVDR. Retrieved from <a href="https://www.gov.uk/guidance/medical-devices-eu-regulations-for-mdr-and-ivdr" target="_blank">https://www.gov.uk/guidance/medical-devices-eu-regulations-for-mdr-and-ivdr</a> [accessed 21 June 2018]
European Commission. (2018). The new Regulations on medical devices. Retrieved from <a href="https://ec.europa.eu/growth/sectors/medical-devices/regulatory-framework_en" target="_blank">https://ec.europa.eu/growth/sectors/medical-devices/regulatory-framework_en</a> [accessed 21 June 2018]
Medicines and Healthcare products Regulatory Agency. (2017). Medical devices: EU regulations for MDR and IVDR. Retrieved from <a href="https://www.gov.uk/guidance/medical-devices-eu-regulations-for-mdr-and-ivdr" target="_blank">https://www.gov.uk/guidance/medical-devices-eu-regulations-for-mdr-and-ivdr</a> [accessed 23 June 2018]
Medicines & Healthcare products Regulatory Agency. (2017). Medical devices: the regulations and how we enforce them. Retrieved from <a href="https://www.gov.uk/government/publications/report-a-non-compliant-medical-device-enforcement-process/how-mhra-ensures-the-safety-and-quality-of-medical-devices" target="_blank">https://www.gov.uk/government/publications/report-a-non-compliant-medical-device-enforcement-process/how-mhra-ensures-the-safety-and-quality-of-medical-devices</a> [accessed 21 June 2018]
Payne, J. (2014). Scottish Parliament information Centre Briefing for the Public Petitions Committee (PE01517). Retrieved from <a href="http://www.parliament.scot/ResearchBriefingsAndFactsheets/Petitions%20briefings%20S4/PB14-1517.pdf" target="_blank">http://www.parliament.scot/ResearchBriefingsAndFactsheets/Petitions%20briefings%20S4/PB14-1517.pdf</a> [accessed 2018 June 28]
In discussion with a representative from Health Facilities Scotland. (2018).
National Cancer Institute. (2008). Hand holding a breast implant which is a flexible sac filled with silicone gel [Accessed 23rd May 2018. Retrieved from <a href="Wikimedia Commons" target="_blank">Wikimedia Commons</a> [accessed 23 May 2018]
SPICe. (2010). Briefing for the Public Petitions Committee (PE1378). Retrieved from <a href="http://www.scottish.parliament.uk/ResearchBriefingsAndFactsheets/Petitions%20briefings%20S3/PB10-1378.pdf" target="_blank">http://www.scottish.parliament.uk/ResearchBriefingsAndFactsheets/Petitions%20briefings%20S3/PB10-1378.pdf</a> [accessed 23 May 2018]
Martindale, V., & Menache, A. (2013). The PIP scandal: an analysis of the process of quality control that failed to safeguard women from the health risks. Journal of the Royal Society of Medicine, 106(5), 173-177. doi: https://doi.org/10.1177/0141076813480994
Berry, M.G., & Stanek, J.J. (2012). The PIP mammary prosthesis: a product recall study. Journal of Plastic, Reconstructive & Aesthetic Surgery, 65(6), 697-704. doi: https://doi.org/10.1016/j.bjps.2012.02.019
The Telegraph. (2012). Breast implant scandal: timeline of how events unfolded. Retrieved from <a href="https://www.telegraph.co.uk/news/health/news/9146338/Breast-implant-scandal-timeline-of-how-events-unfolded.html" target="_blank">https://www.telegraph.co.uk/news/health/news/9146338/Breast-implant-scandal-timeline-of-how-events-unfolded.html</a> [accessed 28 June 2018]
Department of Health and Social Care. (2013). PIP Silicone Breast Implants: Review of the actions of the MHRA and Department of Health. Retrieved from <a href="https://www.gov.uk/government/publications/pip-silicone-breast-implants-review-of-the-actions-of-the-mhra-and-department-of-health" target="_blank">https://www.gov.uk/government/publications/pip-silicone-breast-implants-review-of-the-actions-of-the-mhra-and-department-of-health</a> [accessed 20 June 2018]
Department of Health. (2012). Poly Implant Prothèse (PIP) silicone breast implants Review of the actions of the Medicines and Healthcare products Regulatory Agency (MHRA) and Department of Health. Retrieved from <a href="Poly Implant Prothèse (PIP) silicone breast implants Review of the actions of the Medicines and Healthcare products Regulatory Agency (MHRA) and Department of Health" target="_blank">Poly Implant Prothèse (PIP) silicone breast implants Review of the actions of the Medicines and Healthcare products Regulatory Agency (MHRA) and Department of Health</a> [accessed 20 June 2016]
The Lancet. (2009). Hand holding PIP breast implant. Retrieved from <a href="Doi: https://doi.org/10.1016/S0140-6736" target="_blank">Doi: https://doi.org/10.1016/S0140-6736</a> [accessed 29 May 2018]
National Institutes of Health. (2006). An anteroposterior (A-P) X-ray of a pelvis showing a total hip joint replacement. Retrieved from <a href="Wikimedia Commons" target="_blank">Wikimedia Commons</a> [accessed 23 May 2018]
National Join Registry. (2009). National Joint Registry for England and Wales 6th Annual Report. Retrieved from <a href="http://www.njrcentre.org.uk/NjrCentre/Portals/0/Sixth%20annual%20NJR%20report.pdf" target="_blank">http://www.njrcentre.org.uk/NjrCentre/Portals/0/Sixth%20annual%20NJR%20report.pdf</a> [accessed 20 June 2018]
DePuySynthes. (2009). News and Press. Retrieved from <a href="https://www.depuysynthes.com/about/news-press" target="_blank">https://www.depuysynthes.com/about/news-press</a> [accessed 20 June 2018]
Medicines and Healthcare products Regulatory Agency. (2012). Medical Device Alert Ref: MDA/2012/036 Issued: 25 June 2012 at 11:00. Retrieved from <a href="file:///C:/temp/MDA-2012-036_Archived.pdf" target="_blank">file:///C:/temp/MDA-2012-036_Archived.pdf</a> [accessed 20 June 2018]
Medicines and Healthcare products Regulatory Agency. (2017). All metal-on-metal (MoM) hip replacements: updated advice for follow-up of patients. Retrieved from <a href="https://www.gov.uk/drug-device-alerts/all-metal-on-metal-mom-hip-replacements-updated-advice-for-follow-up-of-patients" target="_blank">https://www.gov.uk/drug-device-alerts/all-metal-on-metal-mom-hip-replacements-updated-advice-for-follow-up-of-patients</a> [accessed 20 June 2018]
The Telegraph. (2017). Hip Implant Maker Alerted Safety Fears Surgeon Told Borders. Retrieved from <a href="https://www.telegraph.co.uk/news/2017/04/11/hip-implant-maker-alerted-safety-fears-surgeon-told-borders/" target="_blank">https://www.telegraph.co.uk/news/2017/04/11/hip-implant-maker-alerted-safety-fears-surgeon-told-borders/</a> [accessed 23 May 2018]
Cohen, D. (2011). Out of joint: the story of the ASR. BMJ, 342(2905). doi: https://doi.org/10.1136/bmj.d2905
British Hip Society. (2012). British Hip Society Newsletter 2012. Retrieved from <a href="https://www.britishhipsociety.com/uploaded/Newsletter%20July%202012.pdf" target="_blank">https://www.britishhipsociety.com/uploaded/Newsletter%20July%202012.pdf</a> [accessed 20 June 2018]
House of Commons Library. (2018). Briefing Paper: Surgical mesh implants. Retrieved from <a href="file:///C:/temp/CBP-8108%20(1).pdf" target="_blank">file:///C:/temp/CBP-8108%20(1).pdf</a> [accessed 20 June 2018]
Heneghan, C. (2017). Transvaginal Mesh Timeline. Retrieved from <a href="https://www.cebm.net/2017/12/transvaginal-mesh-timeline/" target="_blank">https://www.cebm.net/2017/12/transvaginal-mesh-timeline/</a> [accessed 23 May 2018]
Polypropylene Mesh Adjust Single - Incision Mid Urethal Sling. (n.d.) Retrieved from <a href="Creative Commons" target="_blank">Creative Commons</a> [accessed 23 May 2018]
BBC News. (2014). Scottish Health Secretary Alex Neil requests mesh implant suspension. Retrieved from <a href="https://www.bbc.co.uk/news/uk-scotland-scotland-politics-27884794" target="_blank">https://www.bbc.co.uk/news/uk-scotland-scotland-politics-27884794</a> [accessed 20 June 2018]
Chartered Society of Physiotherapy. (2014). Suspension of mesh surgery in Scotland means women need improved access to physiotherapy. Retrieved from <a href="http://www.csp.org.uk/press-releases/2014/08/18/suspension-mesh-surgery-scotland-means-women-need-improved-access-physioth" target="_blank">http://www.csp.org.uk/press-releases/2014/08/18/suspension-mesh-surgery-scotland-means-women-need-improved-access-physioth</a> [accessed 20 June 2018]
BBC News. (2017). Mesh expert voices concerns over patient safety. Retrieved from <a href="https://www.bbc.co.uk/news/uk-scotland-41408704" target="_blank">https://www.bbc.co.uk/news/uk-scotland-41408704</a> [accessed 20 June 2018]
The Scottish Government. (2017). The Scottish Independent Review of the Use, Safety and Efficacy of Transvaginal Mesh Implants in the Treatment of Stress Urinary Incontinence and Pelvic Organ Prolapse in Women: Final Report. Retrieved from <a href="http://www.gov.scot/Publications/2017/03/3336" target="_blank">http://www.gov.scot/Publications/2017/03/3336</a> [accessed 23 May 2018]
House of Commons Library. (2018). Debate Pack: Surgical Mesh. Retrieved from <a href="https://www.parliament.uk/documents/commons-library/Surgical-mesh.pdf" target="_blank">https://www.parliament.uk/documents/commons-library/Surgical-mesh.pdf</a> [accessed 20 June 2018]
BBC News. (2018). Review ordered into epilepsy drug, vaginal mesh and Primodos. Retrieved from <a href="https://www.bbc.co.uk/news/health-43143319" target="_blank">https://www.bbc.co.uk/news/health-43143319</a> [accessed 23 June 2018]
Life Sciences in Scotland. (2018). Medical Technologies. Retrieved from <a href="https://www.lifesciencesscotland.com/key-subsectors/med-tech" target="_blank">https://www.lifesciencesscotland.com/key-subsectors/med-tech</a> [accessed 29 June 2018]
Nuclear Decommissioning Authority. (2014). Guidance on Technology Readiness Levels. Retrieved from <a href="https://www.gov.uk/government/news/guidance-on-technology-readiness-levels" target="_blank">https://www.gov.uk/government/news/guidance-on-technology-readiness-levels</a> [accessed 13 June 2018]
Health Innovation Assessment Portal. (2013). Generating Ideas to Support the NHS. Retrieved from <a href="http://www.hiap-scotland.org/" target="_blank">http://www.hiap-scotland.org/</a> [accessed 21 June 2018]
PwC. (2011). Representation of a Medical Device Innovation. Retrieved from <a href="http://usblogs.pwc.com/emerging-technology/powering-the-innovation-life-cycle/" target="_blank">http://usblogs.pwc.com/emerging-technology/powering-the-innovation-life-cycle/</a> [accessed 23 June 2018]
Healthcare Innovation Assessment Portal. (2015). HIAP-Scotland: Innovators’ Guide. Retrieved from <a href="http://www.hiap-scotland.org/Content/documents/HIAP-Scotland_Innovators_Guide.pdf" target="_blank">http://www.hiap-scotland.org/Content/documents/HIAP-Scotland_Innovators_Guide.pdf</a> [accessed 26 June 2018]
MHRA. (2017). Medicines and Healthcare Products Regulatory Agency Annual Report and Accounts 2016 to 2017. Retrieved from <a href="https://www.gov.uk/government/publications/medicines-and-healthcare-products-regulatory-agency-annual-report-and-accounts-2016-to-2017" target="_blank">https://www.gov.uk/government/publications/medicines-and-healthcare-products-regulatory-agency-annual-report-and-accounts-2016-to-2017</a>
Wright, M.S. (2016). A Case for Randomized, Double-Blinded, Sham-Controlled Class III Medical Device Trials. Yale Law and Policy Review, 34(1). doi: http://digitalcommons.law.yale.edu/ylpr/vol34/iss1/7
NHS National Services Scotland. (2016). National Procurement Interim Strategy. Retrieved from <a href="http://www.train2procure.scot.nhs.uk/supportingdocuments/2016-12-22%20NP%20Procurement%20CoE%20Strategy%20V1.pdf" target="_blank">http://www.train2procure.scot.nhs.uk/supportingdocuments/2016-12-22%20NP%20Procurement%20CoE%20Strategy%20V1.pdf</a> [accessed 21 June 2018]
In discussion with a representative from Medical Devices Unit (Greater Glasgow and Clyde). (2018).
Brown, A. (2017). Procurement Transformation Programme: Working for Scotland’s Patients. Retrieved from <a href="file:///C:/Users/c677140/Downloads/Procurement%20Transformation%20Programme%20-%20Working%20for%20Scotland%E2%80%99s%20Patients%20(2).pdf" target="_blank">file:///C:/Users/c677140/Downloads/Procurement%20Transformation%20Programme%20-%20Working%20for%20Scotland%E2%80%99s%20Patients%20(2).pdf</a> [accessed 26 June 2018]
Kramer, D.B., Tan, Y.T., Sato, C., & Kesselheim, A.S. (2013). Postmarket surveillance of medical devices: a comparison of strategies in the US, EU, Japan, and China. PLoS medicine, 10(9). doi: https://doi.org/10.1371/journal.pmed.1001519
LNE/G-MED. (2017). Post-Market Surveillance (PMS) requirements under the new European Medical Device Regulations. Retrieved from <a href="https://lne-america.com/library/news/post-market-surveillance-pms-requirements-under-the-new-european-medical-device-regulations" target="_blank">https://lne-america.com/library/news/post-market-surveillance-pms-requirements-under-the-new-european-medical-device-regulations</a> [accessed 20 June 2018]
In discussion with a representative from NHS Tayside. (2018).
Barton, A.J. (2012). The regulation of mobile health applications. BMC Medicine, 10(06). doi: https://doi.org/10.1186/1741-7015-10-46
Burgess, M. (2018). What is the Internet of Things? WIRED explains. Retrieved from <a href="http://www.wired.co.uk/article/internet-of-things-what-is-explained-iot" target="_blank">http://www.wired.co.uk/article/internet-of-things-what-is-explained-iot</a> [accessed 21 June 2018]
Health and Sport Committee. (2018). 9 th Meeting 2018, Session 5. Retrieved from <a href="http://www.parliament.scot/parliamentarybusiness/report.aspx?r=11429&mode=pdf" target="_blank">http://www.parliament.scot/parliamentarybusiness/report.aspx?r=11429&mode=pdf</a> [accessed 20 June 2018]
Medicines and Healthcare products Regulations Agency. (2018). Follow up to Health Committee Oral evidence: Brexit – medicines, medical devices and substances of human origin. Retrieved from <a href="http://data.parliament.uk/writtenevidence/committeeevidence.svc/evidencedocument/health-and-social-care-committee/brexit-the-regulation-of-medicines-medical-devices-and-substances-of-human-origin/written/79324.html" target="_blank">http://data.parliament.uk/writtenevidence/committeeevidence.svc/evidencedocument/health-and-social-care-committee/brexit-the-regulation-of-medicines-medical-devices-and-substances-of-human-origin/written/79324.html</a> [accessed 20 June 2018]